SysTooth (Systems tool for Tooth gene discovery)
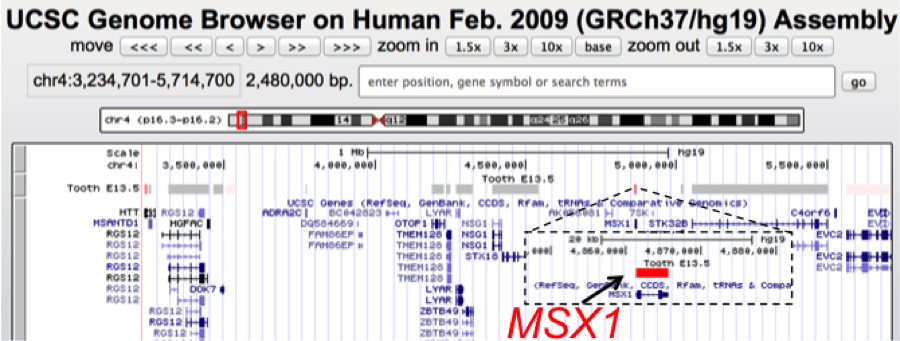
UCSC genome browser view of a ~2.5 Mb genomic interval containing MSX1, a gene associated with tooth agenesis and orofacial clefts. Using SysTooth tracks, we can readily visualize that MSX1 has the highest tooth specific expression among all the genes in this mapped genomic interval and therefore is identified as the best candidate for further analysis. MSX1 has now been associated with several cases of tooth agenesis.
Introduction
Tooth agenesis – one or more missing teeth at birth – is a common birth defect with incidence of 1/10 in certain populations. However, our knowledge of genetic mutations associated with these structural birth defects is limited.
The identification of genetic mutations is traditionally based on linkage analysis and sequencing of candidate genomic regions in patient tissue or animal model. This is typically time-consuming and labor-intensive, and prioritization of candidate genes within an interval is often tricky. Even with the advent of sequencing, prioritization of the select few causative changes among the numerous variations identified is challenging.
Human structural birth defects are often caused by genetic changes that lead to aberrant developmental processes, and therefore it has been proposed that cell or tissue-specific gene expression profiling may facilitate identification of the underlying mutations. Although gene expression datasets for several tissue and cell types currently exist, their application to disease gene discovery has been limited. This is primarily because such datasets are large and prioritization of candidate genes is not simple, especially in the context of development wherein clear control versus mutant comparisons are absent. Similarly, several gene expression atlases, constructed based on in situ hybridization (e.g. http://bite-it.helsinki.fi/), provide insights into developmental gene expression, but such information is not quantitative and does not allow an easy comparison of relative gene expression.
We hypothesize that innovative processing of expression datasets of specific tissues in embryonic development of mammalian models will allow the identification of human disease-associated genes. Based on this principle, we have developed a strategy wherein tissue or cell-specific microarray datasets are subjected to in silico subtraction with a embryonic whole body (WB) reference dataset, which allows the systematic ranking of genes based on their tissue enrichment. To make this tool available to the community, we have developed a web-based public resource termed SysTooth (Systems tool for Tooth gene discovery) that allows efficient identification of genes associated with congenital tooth agenesis.
What is SysTooth based on?
SysTooth presently utilizes microarray gene expression profiles of the E13.5 mouse embryonic molar tooth germ. We identified differentially regulated genes by comparing E13.5 tooth microarray profiles to those representing whole embryonic body (WB). These were then utilized to generate a ranked list of tooth-enriched genes, which can be viewed as SysTooth tracks in the UCSC Genome browser to aid identification of genes with function associated with tooth biology. We performed laser capture microdissection (LCM) to capture mouse embryonic E13.5 tooth germ tissue and then extracted sufficient total RNA to perform microarrays after two rounds of in vitro transcription-based amplification (double amplification). Using the same amplification protocol, we also generated a microarray dataset from total RNA extracted and pooled in equimolar ratios from mouse whole body (WB) tissue at E11.5, E12.5 and E13.5. The tooth specific profiles were “subtracted” from the WB control using a moderated t-test and a tooth enrichment p-value was assigned to each gene. T-statistics were used to rank genes for tooth enrichment.
The significance of in silico subtraction using WB reference dataset
This WB-based in silico subtraction strategy is analogous to the cDNA library screen, based on the significantly higher molar concentration of important transcripts in a specific cell or tissue of interest, as compared to the WB. This is based on the principle that a transcript with high expression in a small organ or cell/tissue (where its expression represents high molar concentration) can be distinguished from basal (or low) level expression of the same transcript in the WB (where its expression represents relatively low molar concentration). Indeed, transcripts with low absolute expression in a specific cell/tissue (compared to WB) can still be expressed at higher molar concentrations tissue-specifically and can thus be identified as enriched, independent of tissue size. Therefore, this strategy does not depend on just raw expression but rather on “exclusive” or “enriched” expression. Hence transcripts that are expressed at lower levels than housekeeping genes, but are highly enriched (high molarity in tissue of interest vs WB), can be effectively identified.
How well does SysTooth work for tooth gene discovery?
We tested the utility of SysTooth to identify genes associated with tooth development and human tooth–related birth defects. These analyses demonstrate that the top 200 highly ranked genes after WB subtraction were highly enriched for genes relevant to tooth biology and odontogenesis, and not for gene encoding housekeeping factors. As expected, the top 200 tooth-enriched genes from the WB subtraction dataset contained genes associated with syndromic and non-syndromic tooth agenesis. This demonstrates that the in silico subtraction strategy which is the basis of SysTooth can successfully identify genes associated with tooth development and disease.
Use SysTooth to prioritize genes associated with tooth development or within a mapped interval for tooth agenesis
SysTooth tracks allow the visualization of genes in context of their enrichment of expression in the tooth. After opening the browser for the human genome assembly window by clicking on these links, the user can type in the interval of interest and the entire genome track is loaded. This representation allows immediate visual detection of the best candidates in a given genomic interval, and allows one to zoom in or out to visualize the presence of promising candidates within a particular region or proximal to it.
To test a genomic region of interest for tooth agenesis candidate genes, open a specific genome assembly on the UCSC Genome Browser with SysTooth tracks by clicking on the following links:
Future: Developing a comprehensive version of SysTooth
Future versions of SysTooth will incorporate extensive microarray data generated from newly generated or published datasets. In addition, by integrating knowledge derived from other genome-wide studies e.g. ToothCODE (http://compbio.med.harvard.edu/ToothCODE/), newer versions of SysTooth will lead to a comprehensive understanding of tooth development and facilitate discovery of genes in genomic intervals or association studies in human cases of tooth-related defects.
Salil Lachke, Ph.D.
Assistant Professor
Department of Biological Sciences
Center for Bioinformatics and Computational Biology
University of Delaware
Newark, DE 19716
E-mail: salil@udel.edu