SysCLFT(Systems tool for Cleft Lip/palate gene discovery)
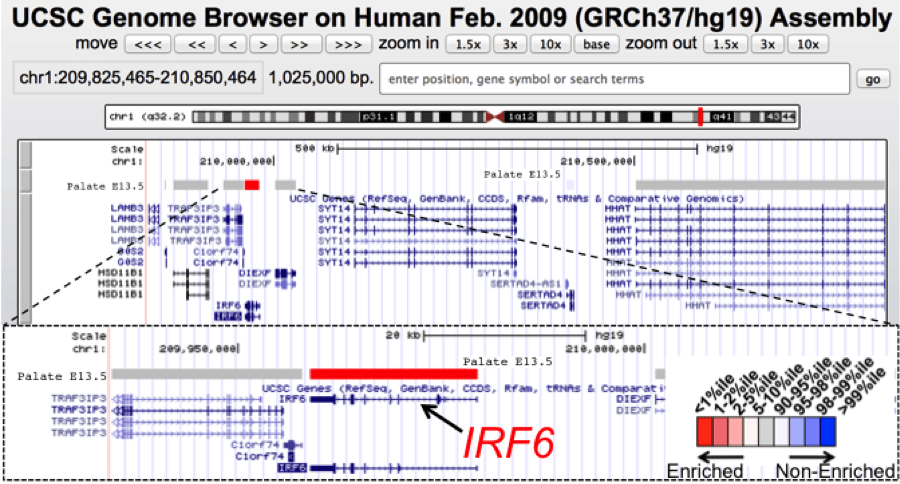
UCSC genome browser view of a 1 Mb genomic interval containing IRF6, a gene associated with orofacial clefts. IRF6 mutations lead to the autosomal dominant van der Woude syndrome (VWS) or the related popliteal pterygium syndrome. VWS includes cleft lip and palate as a prominent feature along with dental anomalies and lip fistulas. Positional cloning of VWS gene (IRF6) took considerable effort with sequencing of many candidate genes in the genomic interval. Using SysCLFT tracks, we can readily visualize that IRF6 has the highest palate specific expression among all the genes in this mapped genomic interval and therefore is identified as the best candidate for further analysis. IRF6 has now been identified as a candidate gene in many genome-wide association studies for non-syndromic orofacial clefts.
Introduction
Orofacial clefts are one of the most common birth defects in the U.S., occurring in 1/800 live-births with a lifetime cost for medical treatment, educational services and lost productivity of more than $100,000 per affected person. Orofacial clefts can occur as primary phenotype – classified as “isolated” or “non-syndromic” – or as one among several phenotypes in a syndrome. There are over 400 syndromic forms of orofacial clefting, however, efforts thus far using linkage analyses have only successfully identified around 40 loci. Moreover, the genetic etiology of non-syndromic manifestation of these malformations has remained largely elusive. Thus a large deficit exists in our knowledge of genes that are associated to these structural birth defects.
The identification of genetic mutations is traditionally based on linkage analysis and sequencing of candidate genomic regions in patient tissue or animal model. This is typically time-consuming and labor-intensive, and prioritization of candidate genes within an interval is often tricky. Since many human genetic diseases are caused by aberrant developmental processes, we hypothesize that innovative processing of expression datasets in embryonic development of mammalian models will allow the identification of human disease-associated genes.
Based on this principle, we have developed an effective strategy to prioritize candidate disease associated genes based on microarray gene expression profiling on embryonic tissues. To make this tool available to the community, we have developed a web-based public resource termed SysCLFT (Systems tool for Cleft Lip/palate gene discovery) that allows efficient identification of genes associated with congenital orofacial clefts.
What is SysCLFT based on?
SysCLFT is based on a strategy termed “in silico subtraction” which is analogous to the principle of identification of cell type enriched genes by cDNA subtraction. SysCLFT presently utilizes microarray gene expression profiles of existing FaceBase mouse embryonic isolated craniofacial (CF) tissue datasets (palate, maxillary, mandible, and frontonasal). We also generated a microarray dataset from total RNA extracted and pooled in equimolar ratios from mouse whole body (WB) tissue at E10.5, E11.5 and E12.5. We identified differentially regulated genes by comparing CF microarray profiles to those representing WB. The CF specific profiles were “subtracted” from the WB control using a moderated t-test and a CF-tissue enrichment p-value was assigned to each gene. t-statistics were used to rank genes for CF enrichment. These were then utilized to generate a ranked list of CF-enriched genes, which can be viewed as SysCLFT tracks in the UCSC Genome browser to aid identification of genes with function associated with CF biology and morphogenesis.
 The significance of using WB reference datasets for in silico subtraction
The significance of using WB reference datasets for in silico subtraction
WB datasets as described above are comprised of multiple developing tissues and thus represent ideal “basal” gene expression profile datasets, Therefore, comparison of tissue-specific profiles against the WB control facilitates identification of tissue-specific gene expression. In a different study, we have shown that an in silico subtracted mouse lens database is an elegant tool to identify lens-enriched genes (https://bioinformatics.udel.edu/research/isyte/) that play key roles in lens biology, and for identification and prioritization of potential candidate genes harboring mutations at mapped human cataract loci. This is consistent with the idea that selective gene expression in a tissue may be reflective of a function in the development or function of the tissue.
How well does SysCLFT work for orofacial cleft gene discovery?
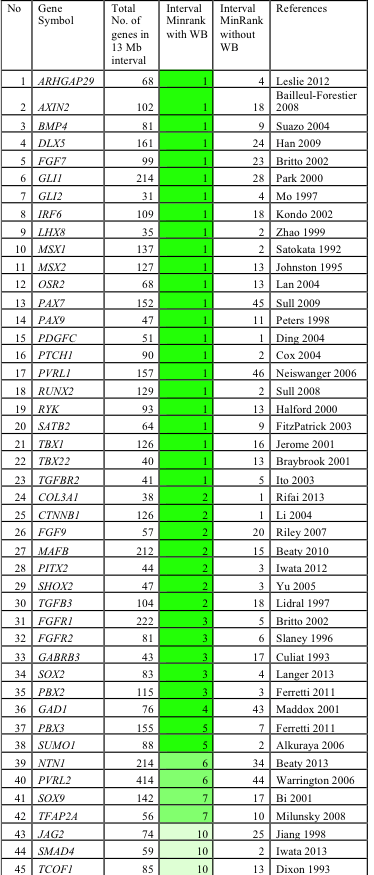
We tested the utility of SysCLFT to identify genes associated with face development and human craniofacial defects. These analyses demonstrate that the top 500 highly ranked genes after WB subtraction were enriched for candidate genes relevant to face and palate development, without being enriched for genes encoding miscellaneous house keeping factors. Interestingly, 41 of 45 genes (91%) linked to CF morphogenesis are identified at higher expression ranks with WB-subtraction compared to only 4 of 45 (9%) without WB-subtraction, highlighting effectiveness of this approach. Specifically, majority of genes with established role in CF development (e.g. Fgf7, Irf6, Pax7, Pax9, Pbx2, Pbx3, Msx1, Runx2, Satb2, Tbx22, etc.) were highly enriched in WB-subtracted datasets from different facial tissues. Most significantly, we find that in silico subtraction successfully identifies a majority (85%, n=45) of clefting and CF candidate genes within the top 5 minRank genes in mean chromosomal intervals of 13Mb, each containing 106 genes on average (Table 1).
These data demonstrate that our in silico subtraction method: 1) can be successfully applied to isolated CF tissues (palate, maxillary, mandible, frontonasal), 2) specifically identifies genes involved in CF development regardless of high or low absolute expression, while filtering out genes with high expression that are not CF tissue-specific (housekeeping genes), and 3) significantly improves prioritization of potential disease genes.
Using SysCLFT for prioritization of genes within a mapped interval for orofacial clefts
SysCLFT tracks allow the visualization of genes in context of their enrichment of expression in CF tissue. After opening the browser for the human genome assembly window, the user can type in the interval of interest and the entire genome track is loaded. This representation allows immediate visual detection of the best candidates in a given genomic interval, and allows one to zoom in or out to visualize the presence of promising candidates within a particular region or proximal to it.
To test a genomic region of interest for orofacial cleft candidate genes, open a specific genome assembly on the UCSC Genome Browser with SysCLFT tracks by clicking on the following links:
- Human hg19 All
- Human hg19 Mandible
- Human hg19 Maxilla
- Human hg19 Frontonasal
- Human hg19 Palate
- Mouse mm9 All
- Mouse mm9 Mandible
- Mouse mm9 Maxilla
- Mouse mm9 Frontonasal
- Mouse mm9 Palate
Future: Developing a comprehensive version of SysCLFT
Future versions of SysCLFT will incorporate extensive microarray data generated from newly generated or published datasets. In combination with other genome-wide studies e.g. NIH-supported FaceBase initiative [https://www.facebase.org/], network of validated gene regulatory relationships, and effective use of bioinformatics algorithms, newer versions of SysCLFT will be more comprehensive and lead to the understanding of orofacial development and pathogenesis, as well as for the highly efficient identification of disease associated genes in genomic intervals in human patients. In collaboration with Dr. Joshua Ho (Victor Chang Institute, Sydney, Australia), we are currently assembling a curated evidence-based gene regulatory network for craniofacial morphogenesis (Fig. 1), which we plan to integrate into future versions of SysCLFT.
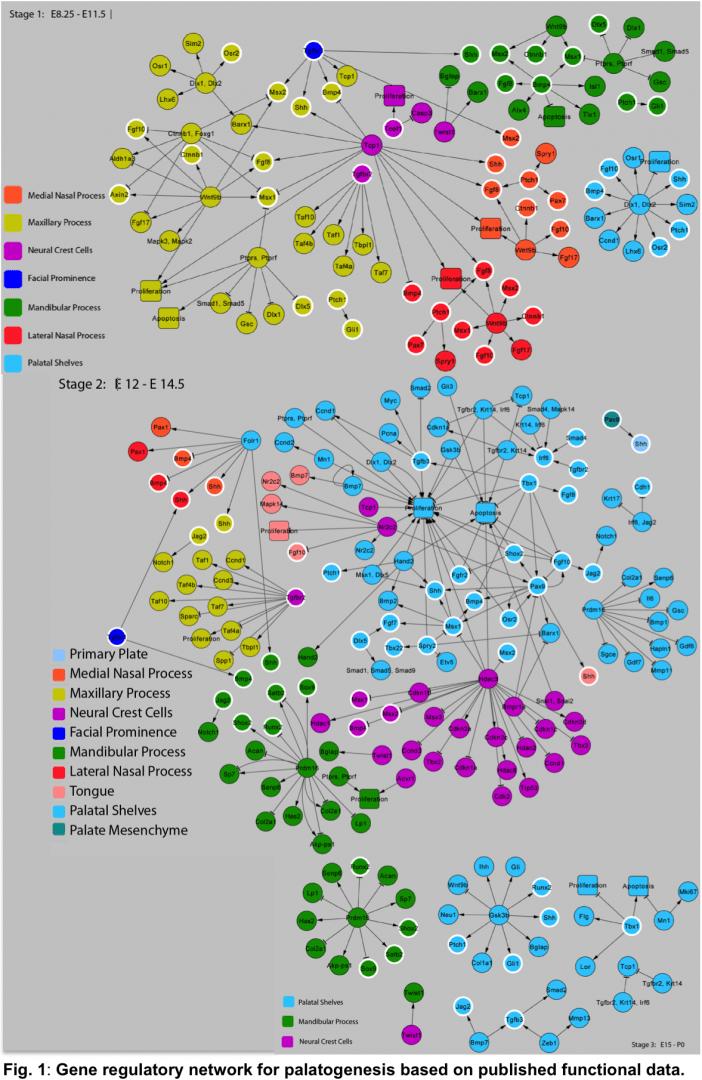
Fig. 1: Gene regulatory network for palatogenesis based on published functional data.
Salil Lachke, Ph.D.
Assistant Professor
Department of Biological Sciences
Center for Bioinformatics and Computational Biology
University of Delaware
Newark, DE 19716
E-mail: salil@udel.edu